Alumna investigadora: Ana Caballo Zulueta
Estudios: Grado en Física. Facultad de Ciencias. Departamento de Electricidad y Electrónica
Profesores/tutores: María Aboy Cebrián, Iván Santos Tejido
Resumen del proyecto:
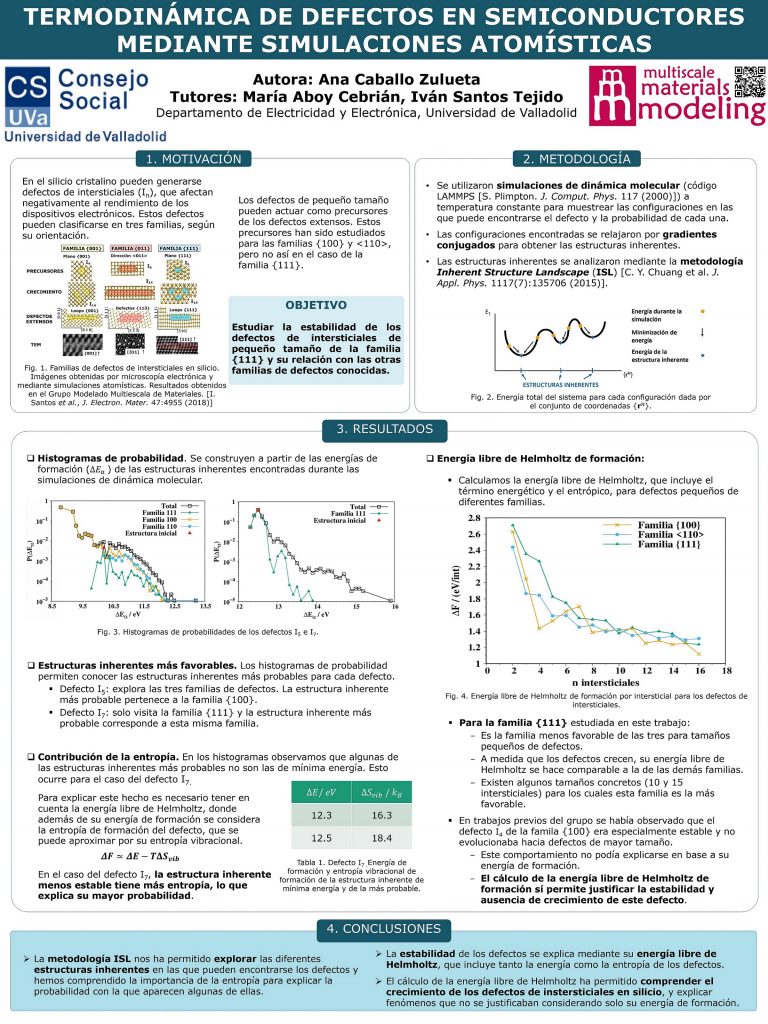
Este proyecto se ha realizado en el Grupo de Modelado Multiescala de Materiales (https://www.ele.uva.es/~mmm/index.html) del Departamento de Electricidad y Electrónica de la Universidad de Valladolid. Mediante simulaciones de dinámica molecular se realizó una caracterización termodinámica de los defectos de intersticiales de la familia {111} que aparecen en silicio cristalino. Esta familia de defectos no había sido estudiada hasta el momento en trabajos previos por ningún autor. Generalmente, la estabilidad de los defectos se estudia atendiendo únicamente a criterios energéticos. Sin embargo, gracias a la metodología Inherent Structure Landscape (ISL) empleada en este trabajo fuimos capaces de evaluar otros factores termodinámicos, como la entropía, que pueden tener gran relevancia.
Sectores de aplicación:
Este tipo de estudios son fundamentales para desarrollar modelos que describan la formación y evolución de defectos durante el procesado de silicio para fabricar dispositivos electrónicos. Por este motivo, las empresas interesadas son las del sector de tecnología del silicio, con las que el grupo en el que se ha realizado esta Beca de Colaboración trabaja frecuentemente.
Objetivos alcanzados:
En primer lugar, hemos podido entender y unificar la metodología ISL, que estaba dispersa en diferentes trabajos de la literatura. Esta metodología nos permitió explorar las diferentes configuraciones en las que pueden encontrarse cada uno de los defectos. Además, identificamos las más favorables, es decir, aquellas que aparecen con mayor frecuencia en las simulaciones de dinámica molecular.
Por otra parte, calculando la energía libre de Helmholtz de formación para diferentes familias y tamaños de defectos, comparamos su estabilidad y justificamos el comportamiento de algunos defectos cuyo crecimiento no había sido explicado anteriormente.
Metodología empleada:
Para la realización del trabajo se utilizaron simulaciones de dinámica molecular a temperatura constante, conocidas como simulaciones de annealing. Para ello se empleó el código de simulación LAMMPS. Los resultados de estas simulaciones se analizaron mediante la metodología ISL, obteniendo así la información de carácter termodinámico requerida.